30.07.2022
Malakazoologen sind diejenigen, die Schnecken bestimmen und untersuchen. Leider sind diese Zoologen bis heute eher Schnecken Leichenfledderer und “Häuslesammler”. Jetzt wollte ich aber unsere einheimischen Schnecken bestimmen und fotografieren, ohne diese irgendwie zu stören.
So kam mir die Idee, nach meinen umfassenden Untersuchungen mit dem Alpenveilchen, bei den Schnecken ebenso mit der DNA zu arbeiten.
Durch Recherche fand ich, dass es eine umfassende Datenbank auf NCBI gibt über 16S als Marker für verschiedene Schnecken (1).
In meinem Labor benutze ich grundsätzlich keine giftigen Chemikalien wie Phenol, Mecaptoethanol oder Chloroform, die man normaler Weise zum Aufreinigen von DNA verwendet. Deshalb habe ich ein Protokoll entwickelt, das für Schnecken funktioniert ohne diese auf irgend eine Weise zu verletzten oder diese Chemikalien zu verwenden.
Dann haben Schnecken viele Polysaccharide, sogenannte Mucopolysaccharide und auch Glycogen. Diese können die PCR stören oder auch die DNA zum scheren bringen. Wenn die DNA geschert ist, erkennt man dies an “Wölkchen” als PCR Produkt. Falls kein PCR Produkt erscheint, ist es auch möglich, dass die DNA nicht in Lösung ging und noch im Eppi klebt.
Ich habe ziemlich viel Zeit in dieses Protokoll investiert und will es hier gerne mit interessierten Menschen teilen.

Extraktion von DNA aus Schnecken ohne Phenol und ohne Chloroform

Ausgangsmaterial ist ein Abstrich mit einem Wattestab (Swab).
Hierbei wird sich die Schnecke zurück ziehen, das macht aber nichts.
Dann kommt der Kopf vom Swab in ein Eppi. Ich schneide oft nur den Teil der Watte ab der mit der Schnecke in Berührung kam. Hier ist aber äußerste Vorsicht geboten, da eine Kontamination mit menschlicher DNA durchaus möglich ist.
Je Eppi nehme ich 800 µl WB Buffer, plus 20 µl DTT, plus 20 µl Proteinase K. Der Puffer mit der Watte kann hier bei -20° Grad weggefroren werden für eine spätere Extraktion.

WB Buffer
100 mM TrisHCl pH 7.6
5 mM EDTA
350 mM Sorbitol
1 % PVP 40
2 % CTAB
2 M Na Cl
Der 1. Schritt ist für manche Schneckenarten wichtig, kann aber erst mal weggelassen werden.
Das Sorbitol verbindet sich mit den Mucopolysacchariden und das PVP Polyvinylpyrrolidone (molweight 40000) entfernt die Polyphenole.
Dies alles wird gut gemischt. Wenn das Eppi mit dem Puffer und der DNA erst aufgetaut werden soll, dann stelle ich dies kurz auf den Heizblock bei 60°C. Dann kommt der Waschschritt:
Das Eppi kommt bei 8000 rpm für 10 Minuten in die Zentrifuge.
Danach schwimmt wohl das Sorbitol mit den Mucopolysacchariden und das PVP mit den Polyphenolen oben und die DNA mit CTAB befindet sich unten.
Also pipettiert man diese oben weg. Dies mache ich nach Gefühl, so etwa die oberen 500 µl der Lösung kommen weg.
Auf die Watte und den unteren Teil kommt dann 800 µl EB, plus 50 µl SDS (10 %) plus 20 µl DTT, plus 20 µl Proteinase K.
EB Buffer

10 mM TrisHCl pH 7.6
10 mM KCl
10 mM MgCl2
400 mM NaCL
2 mM EDTA
1 % CTAB
1 % PVP40
CTAB Puffer löst man am besten über Nacht und nicht zu stark schütteln, schäumt! Das SDS im EB Puffer trübt den Puffer ein, ist aber kein Problem, im Heizblock wird der Puffer wieder klar.
Proteinase K
20 mg/ml in 100mM Tris pH 8,6, 6 mM CaCl2, 50% Glycerin. Lagern bei -20°C.
Proteinase K ist am meisten aktiv bei 65°C mit SDS und sollte mindestens 1x im Jahr neu angesetzt und alliquotiert werden.
DTT
1 g DTT in 6,5 ml H2O oder 1M. Aliquotieren und bei -20°C lagern. DTT oxidiert schnell, deshalb lieber öfters neu machen. DTT ist ein Reduktionsmittel für Disulfidbindungen
Das Eppi mit EB, SDS, DTT, und Proteinase K wird vorsichtig gemischt und kommt bei 60°- 65°C für vier Stunden in den Heizblock. Ab- und an das Eppi bisschen bewegen und mischen, aber nicht zu doll, sonst kann die DNA scheren. Ich hatte es auch schon deutlich länger auf dem Heizblock und danach bei Raumtemperatur (2 Tage), ohne nennenswerte Verluste.

Nach dem Verdau werden 700 µl abpittetiert – diesmal möglichst von unten. Dies kommt mit 600 µl Isopropanol in ein neues Eppi und wird vorsichtig gemischt. Das alles bei Raumtemperatur! Dann wird die DNA 60 Sekunden bei 10 K rpm abzentrifugiert (gerne auch mehr, kann ich aber nicht). Nicht zu lange oder zu kalt, sonst kommt unerwünschtes irgendwas mit oder das Pellet lässt sich nur schwer wieder lösen.
Danach kann man das Pellet mit 70% kaltem EtOH waschen und trocknen. Ich trockne immer ein paar Stunden auf der Bench. Lösen tu ich es mit etwa 50 µl Bidest meist über Nacht auch auf der Bench.
Es ist wichtig eventuelle Reste der Proteinase K zu deaktivieren, da sonst die Taq verdaut wird! Deshalb erhitze ich hier die DNA nochmal etwa 10 Minuten bei 80 °C.
Falls die PCR später nicht funktioniert, kann das am ehesten an der DNA liegen. Natürlich nur, wenn man sich mit den Primern sicher ist. Hier empfiehlt sich eine positiv Kontrolle. Probleme mit der DNA kann z.B. von der Proteinase K kommen. Meiner Erfahrung nach kann man da mit der Konzentration und der Verdauzeit eher hoch gehen, als runter. RNAse benutze ich meistens keine.
Dann wieder die Mucopolysaccharide, Polyphenole und auch Glycogen. Diese können die PCR stören oder auch die DNA zum scheren bringen. Wenn die DNA geschert ist, erkennt man dies an “Wölkchen” als PCR Produkt. Hier muss man nochmal alles neu machen. Vielleicht einen 2. Schritt mit dem WB einfügen. Falls kein PCR Produkt erscheint, ist es auch möglich, dass die DNA nicht in Lösung ging und noch im Eppi klebt.
Note: schnelle Methode!
Nein, genomic DNA kits haben bei mir zu keinem Ergebnis geführt. Nicht mal bei der Schnirkelschnecke, die echt einfach ist. Aber ich will hier ein schnelles Protokoll einfügen, das man zuerst probieren kann:
Als Puffer geht womöglich auch einfacher PCR Puffer mit Proteinase K, SDS und DTT. Verdau möglichst über Nacht. Was ich gerne mache ist, die DNA nach dem Verdau einfach in Roti-Spin MINI-100 (Art. CL-15.1) von Roth. Das kann man da dann mit Bidest waschen und dann in 50 µl H2O aufnehmen. Also mit “einfachen” Tieren funktioniert dies einwandfrei.
Ich möchte nochmals darauf hin weisen, dass Proteinase K relativ frisch sein muss, mit SDS aktiviert wird und DTT oxidieren kann.
Bei mir war meist alte Prot K der Knackpunkt. Am besten alliquotieren und wegfrieren, nach nem halben Jahr kann man das dann meinst auch vergessen. Etwas SDS im Gelladepuffer bringt bessere Banden bei kleinen PCR Produkten.
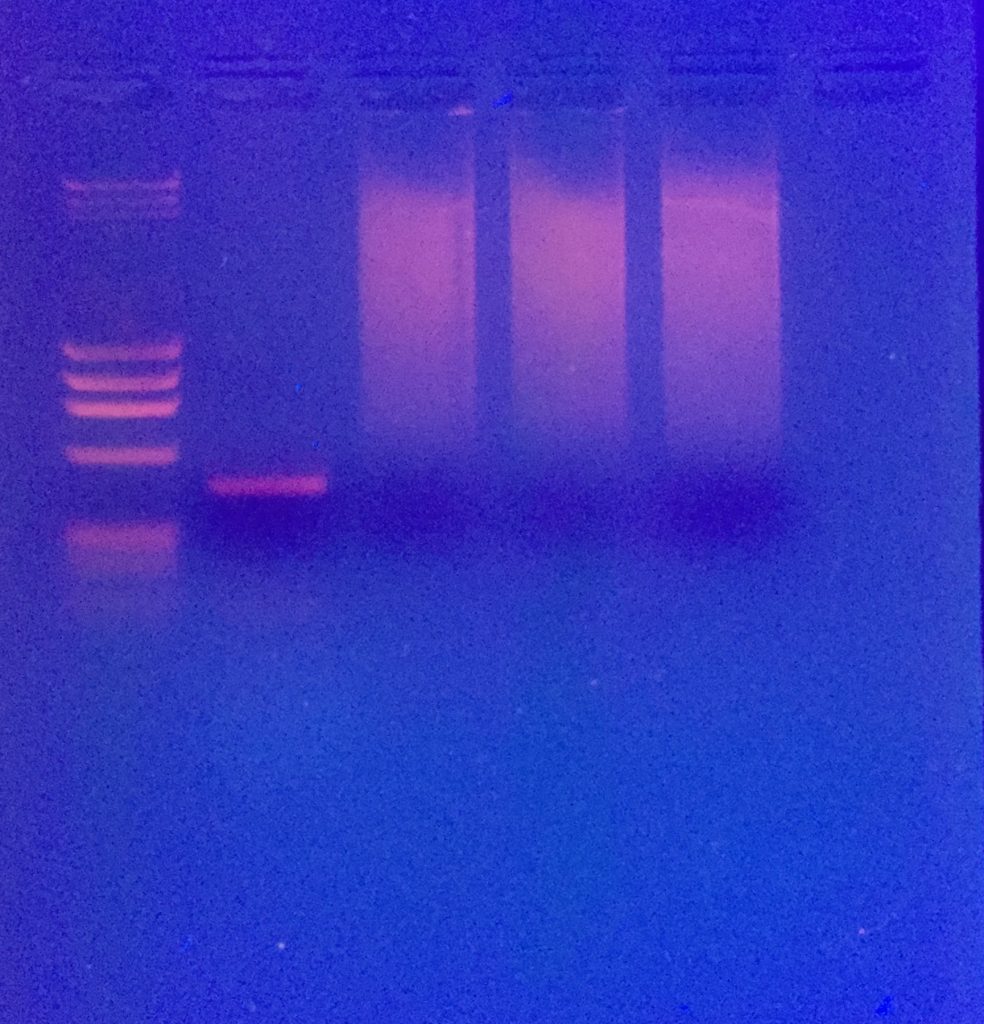
PCR der 16S mitochondrialen rDNA von Schnecken
Die Primer sind alt bekannt und wurden 1991 von Palumbi veröffentlicht (2). Ich habe sie allerdings etwas modifiziert:
HM106f – CGGCCGCCTGTTTATCAAAAACAT
HM107r – GGAGCTCCGGTTTGAACTCAGATC
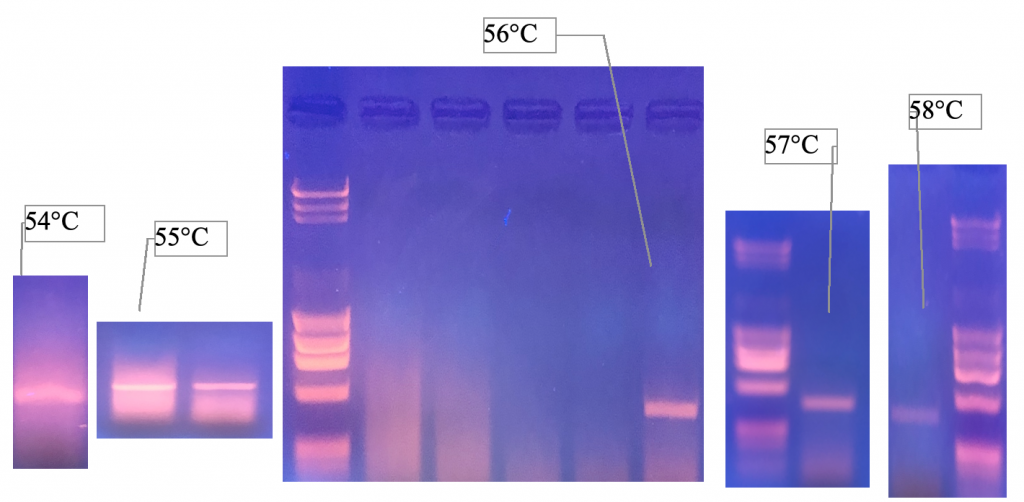
PCR: 30 cycles mit annealing 56°C und Phusion Taq (NEB)
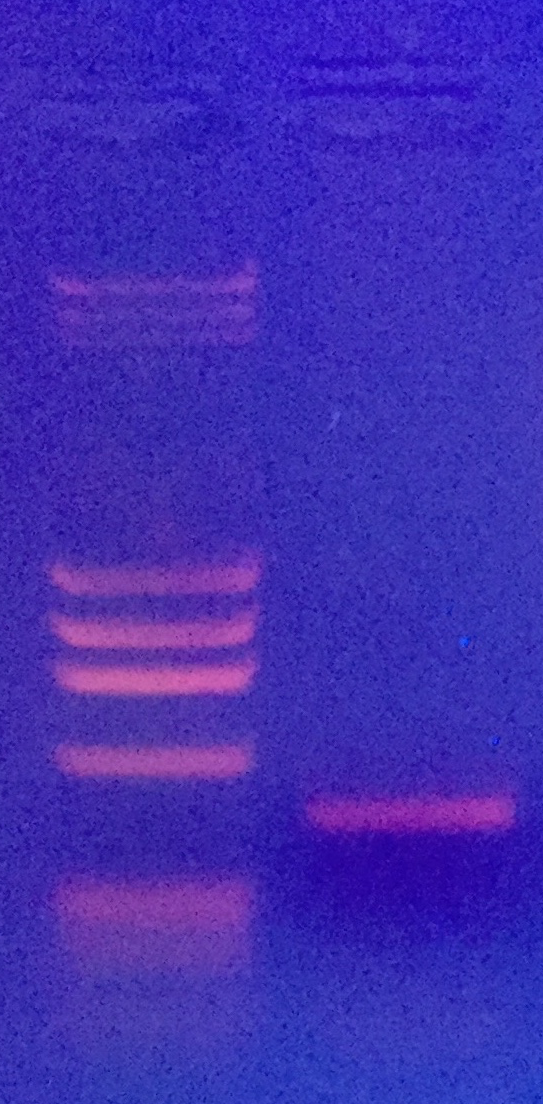
Die Bande wird ausgeschnitten, mit dem Monarch DNA kit von New England Labs aufgereinigt, bei GATC in Köln mit Primer HM103 sequenziert und die Sequenz mit NCBI Blast abgeglichen.
Hier das Ergebnis mit meinem ersten Versuch, der oben abgebildeten Posthornschnecke.

(1) Razkin et al., Molecular phylogeny of the western Palaearctic Helicoidea (Gastropoda, Stylommatophora). Molecular Phylogenetics and Evolution, Volume 83, Pages 99-117 (2015)
(2) Palumbi, S.R., Martin, A., Romano, S., McMillan, W.O., Stice, L., Grabowski, G., The Simple Fool’s Guide to PCR, Version 2.0. Department of Zoology, University of Hawaii, Honolulu, HI. (1991)
(3) Jean-René Arseneau, Royce Steeves, Mark Laflamme, Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Mol Ecol Resour (2017) DOI: 10.1111/1755-0998.12616
(4) Peter W. Inglis, Marilia de Castro R. Pappas, Lucileide V. Resende, Dario Grattapaglia, Fast and inexpensive protocols for consistent extraction of high quality DNA and RNA from challenging plant and fungal samples for high-throughput SNP genotyping and sequencing applications, PLOS one (2018) https://doi.org/10.1371/journal.pone.0206085

Text und Abbildungen dürfen unter den Bedingungen der Creative Commons Attribution Share-Alike license (CC-BY-SA) gerne ganz oder teilweise kopiert und zitiert werden.